████████╗ ██████╗ █████╗ ██╗ ██╗ ██╗ █████╗ ██████╗
╚══██╔══╝ ██╔══██╗ ██╔══██╗ ██║ ██╔╝ ██║ ██╔══██╗ ██╔══██╗
██║ ██████╔╝ ███████║ █████╔╝ ██║ ███████║ ██████╔╝
██║ ██╔═══╝ ██╔══██║ ██╔═██╗ ██║ ██╔══██║ ██╔══██╗
██║ ██║ ██║ ██║ ██║ ██╗ ███████╗ ██║ ██║ ██████╔╝
╚═╝ ╚═╝ ╚═╝ ╚═╝ ╚═╝ ╚═╝ ╚══════╝ ╚═╝ ╚═╝ ╚═════╝
to accelerate research
on infectious disease
Welcome to the homepage of the computational lab of Ted Pak, MD, PhD. We are based in the Division of Infectious Diseases at the University of California, Irvine.
Our goal is to fully leverage machine learning, artificial intelligence, and multiscale data sources to generate higher quality evidence for the management of infectious diseases. Our work spans the fields of clinical informatics, epidemiology, microbial genomics, and computer science. Diseases we have published on include sepsis, COVID-19, and MRSA bacteremia.
We aim to create well-validated models and reusable software that can be deployed across healthcare systems to improve outcomes for patients at risk of infection. Current areas of interest include:
Massive analysis of clinical notes with LLMs
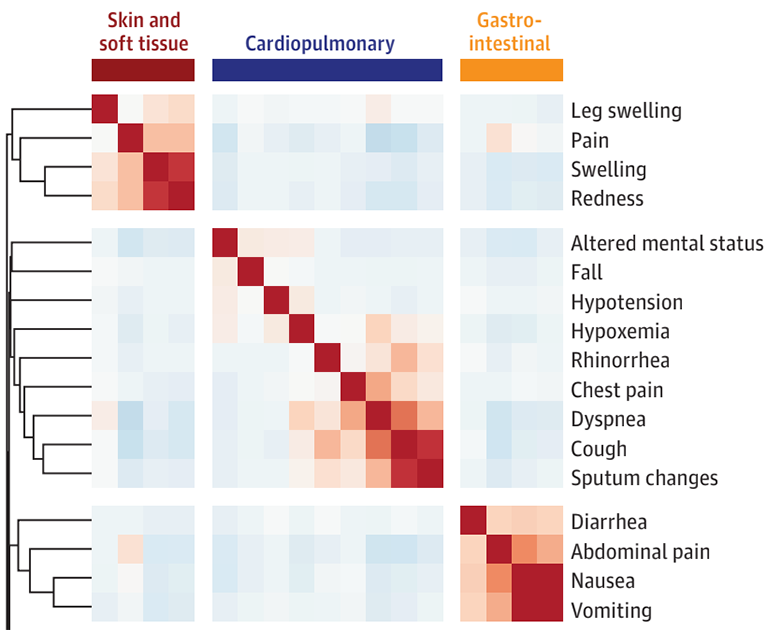
Many questions in infectious diseases are now primarily studied by analysing electronic medical records. Until recently, these studies largely ignored what clinicians wrote in notes in favor of numeric and structured data like lab results. The result is that predictive and statistical models often use very different data than human clinicians—e.g., most models of sepsis ignore the patient's self-reported symptoms.
We've adapted large language models into lightning-fast "chart reviewers" that can scan thousands of clinical notes in seconds. This has allowed us to discover data-driven, symptom-based clusters of sepsis patients that can refine algorithms for antibiotic choice. For more, see Pak et al. JAMA Netw Open 2025.
Genomic and spatiotemporal detection of hospital outbreaks
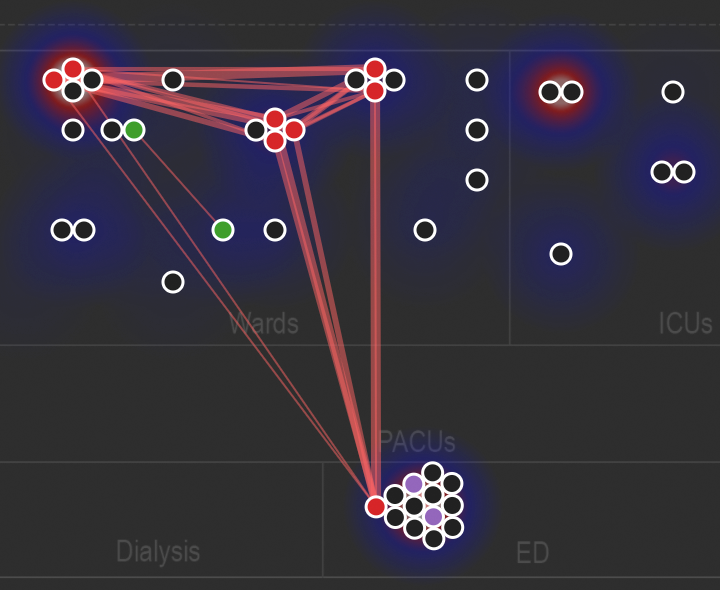
The advent of cheap whole genome sequencing for bacteria, viruses, and other pathogens allows us to characterize the spread of these pathogens within hospitals in great detail. We have developed open source software (see: pathospot.org) that visualizes phylogenies in the context of patient location data to spot hospital outbreaks and narrow down the epidemiologic links for likely transmission events.
Many challenges remain in cost-effectively deploying these technologies for routine prospective surveillance, rather than select outbreak investigations. For instance, MRSA outbreaks can persist for years via long-term colonization. For more, see Berbel Caban et al. Genome Med 2020.
Assessing efficacy of infection prevention strategies
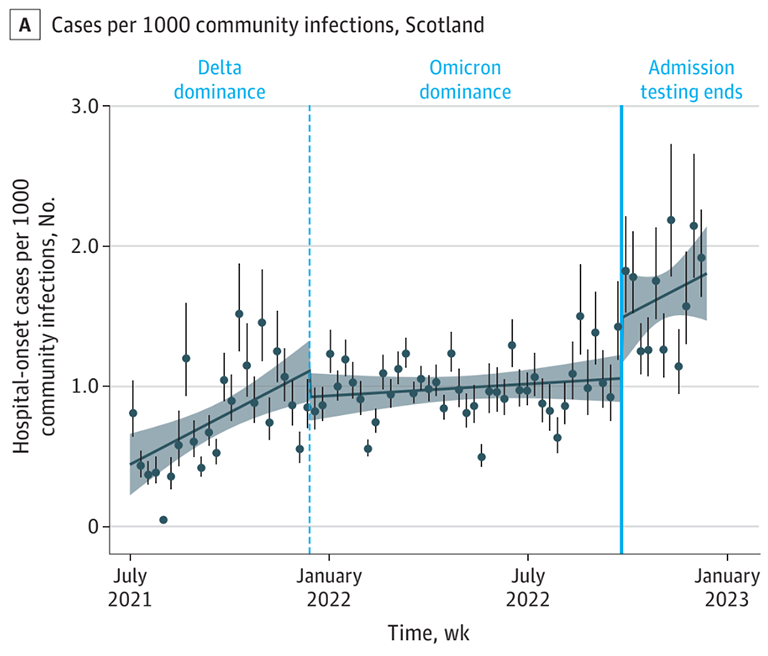
Infection prevention strategies for respiratory viruses in hospitals are unlikely to ever be tested in randomized controlled trials. Even with perfect randomization, it is difficult to implement blinding, placebos, and clinical equipoise for nonpharmaceutical interventions like universal masks and universal testing. There is no broad consensus among US hospitals as to when these precautions should be started and stopped, e.g., during seasonal surges of infections.
We have used comprehensive data from both public health surveillance sources and electronic medical records to model epidemiologic changes surrounding abrupt changes in respiratory viral precautions, estimating their likely efficacy. For more, see Pak et al. JAMA Intern Med. 2023.